2.10 Catalepsy procedure
Mice (n = 3) were initially injected (i.p.) with vehicle (0.9% saline/0.2% lactic acid), 10.0 mg/kg nuciferine, or 1.0 mg/kg haloperidol. Mice were placed upright on a 45° angled screen. The time required for the animal to move all four paws was scored in seconds (maximum of 5 min) and is reported as the latency to movement. An extended delay to move on the inclined screen test is indicative of drug-induced catalepsy. Power analyses were performed with the resulting data. The experiment had 100% power to detect a significant difference (α = 0.05) between nuciferine and haloperidol at the 60 minute timepoint.
2.11 DOI drug discrimination
Adult male NIH Swiss mice (n = 6) were trained to respond under an FR5 reinforcement schedule by presentation of evaporated milk in daily sessions using procedures similar to those previously described. [25] Mice were trained in drug discrimination via injection of saline (VEH) or 0.3 mg/kg R(-)-DOI presented in a pseudo-random order, with the constraint that no animal could receive the same injection for more than 3 consecutive sessions (1 session / day). Response assignments were counterbalanced across trials. Drugs were administered i.p. and pre-treatment time was 10 minutes. During each training session the overall response rate, overall distribution of responses on the drug-injection lever, and the distribution of responses on this same lever prior to delivery of the first reinforcer were analyzed. When animals reliably achieved a level of >85% correct responding prior to delivery of the first reinforcer over 3 consecutive sessions, a substitution test occurred the following day. During test sessions, a multiple component cumulative dosing procedure was used, and no responses were reinforced. Each component was terminated after the emission of five responses on either lever. Mice were then removed from the chamber, administered the next cumulative dose, and returned to the chamber. Ten minutes later, levers were re-extended into the experimental chamber. In this manner, four doses of drug could be tested over ~40 min in a single session. The distribution of responses between the two levers was expressed as a percentage of total responses emitted on the drug-appropriate lever. Response rate was calculated for each session by dividing the total number of responses emitted on both levers by the elapsed time prior to 5 responses on either lever. Nuciferine was administered 15 minutes prior to the first injection of DOI.
2.12 PCP drug discrimination
Adult male Sprague-Dawley rats (n = 5) were trained to respond under an FR20 schedule and were reinforced by presentation of food pellets in daily sessions using procedures similar to those previously described. [26] Rats were trained in drug discrimination via a pre-session injection of saline (VEH) or 3 mg/kg PCP chosen in a pseudo-random order (coin flip), with the same constraints, criteria, and dosing procedures as described above except that during test sessions, a given component of the cumulative dosing procedure was terminated after the emission of 20 responses on either lever. Drugs were administered i.p. and pretreatment time was 10 minutes. As described above for DOI, four doses of drug could be tested in a single ~40 min test session. Nuciferine was administered 15 minutes prior to the first injection of PCP.
2.13 Clozapine drug discrimination study
Adult male B6129 hybrid mice (n = 12) were trained to respond under a FR10 schedule and were reinforced by presentation of sweetened milk as described previously. [27] The drug and vehicle lever positions were counterbalanced between groups to control for olfactory cues. [28] All injections were given subcutaneously with a pre-session injection time of 30 minutes. Training occurred on a double alternation injection schedule with two days of VEH followed by two days of CLZ and repeated (VEH, VEH, CLZ, CLZ, VEH, VEH etc.). In order for a mouse to pass a training day it had to meet three criteria: (1) complete the first fixed ratio (FR) on the condition-appropriate lever, (2) at least 80% of the total responses were made on the condition-appropriate lever, and (3) at least 10 responses per minute were made during the session. Drug testing was conducted approximately two times per week with at least two training days in between. To be eligible for testing, mice were required to pass both a drug and vehicle training day consecutively. During drug substitution tests, animals were injected subcutaneously with nuciferine (0.1, 0.3, 1.0, 3.0, 10.0 mg/kg) and placed in the operant chamber after 30 minutes. Responses on both levers were reinforced.
2.14 Prepulse inhibition (PPI)
PPI of the acoustic startle response was conducted as described elsewhere [29] using SR-LAB chambers (San Diego Instruments, San Diego, CA). To determine whether nuciferine could ameliorate or normalize PPI, two separate experiments were conducted. In the first, C57BL/6J mice were administered VEH, 5, or 10 mg/kg nuciferine (i.p.) and returned to their home-cages for 15 min. Subsequently mice were treated with either VEH or 6 mg/kg PCP (i.p.) and placed into the PPI apparatus for a 5 min habituation prior to the onset of testing. In the second study, WT and DAT-KO mice were given VEH or 2.5, 5, or 10 mg/kg nuciferine or 2 mg/kg clozapine (i.p.) and returned to their home-cages. Fifteen min later DAT mice were habituated to the PPI apparatus for 5 min and testing began. The startle trials consisted of a 40 msec burst of 120dB white-noise; prepulse trials consisted of 20 msec prepulse stimuli that were 4, 8, or 12 dB above the white-noise background (64dB) and were followed 100 msec later by the 120dB acoustic startle stimulus. Non-stimulus or null trials consisted of the 64dB white-noise background. PPI responses were calculated as a percentage score for each prepulse intensity, where %PPI = [1–(prepulse trials/startle-only trials)]*100.
2.15 Statistics
The data are presented as means and standard errors of the mean (SEM) and data from the locomotor activity, head-twitch, and PPI studies were analyzed by SPSS statistical software (IBM Corp., Armonk, NY). Locomotor data were assessed with repeated measures ANOVA (RMANOVA) for within subject effects of time and for between subjects effects of treatment; head-twitch data were similarly assessed with RMANOVA. ANOVA and independent measures t-test were used to also examine treatment differences in motor activity. For the PPI experiment, the responses to the null and startle stimuli were analyzed by two-way ANOVA, whereas the PPI data were subjected to RMANOVA where the within subjects effect was prepulse intensity and the between subjects effects were treatment and, in the case of the DAT mice, genotype. All post-hoc analyses were by Bonferroni corrected pair-wise comparisons. A p<0.05 was considered significant. In the drug discrimination studies for each test session, mean (±SEM) percent responding on the drug-associated lever and the rate of responding (responses/sec) were calculated for each session component. Full substitution was operationally defined as >80% selection of the drug-associated lever, partial substitution was operationally defined as 40%-80% selection of the drug-associated lever, and no substitution was operationally defined as <40% selection of the drug-associated lever. Subjects failing to complete the response requirement in a given component were excluded from the data analyses, but their data were included in response rate calculations. Discrimination and response rate data were not normally distributed, so the Kruskal-Wallis one-way ANOVA on ranks was used to analyze data across dose, comparing 3 treatment conditions: 1) training drug alone, 2) training drug + 1.0 mg/kg nuciferine, and 3) training drug + 3.0 mg/kg nuciferine. Significant ANOVAs were followed by pair-wise multiple procedures using the Dunn's method (α = 0.05) to determine differences among means.
3. Results
3.1 Prediction, in vitro identification, and in vitro characterization of nuciferine
The in silico assessment of phytochemicals in Nelumbo nucifera (Fig 3) suggested that nuciferine and its metabolites are the most structurally similar to known compounds (Fig 3; color of circles). Additionally, nuciferine and its metabolites have high confidence protein-binding predictions (Fig 3; size of circles) and are predicted to cross the blood brain barrier (y axis). Finally, nuciferine is predicted to have a relatively large number of molecular targets (x axis).
Fig 3. Bioinformatic predictions of lotus phytochemicals.

X axis = number of unique predicted targets, as predicted by SEA. Each unique target included all affinity classes. Y axis = prediction of blood brain barrier penetration using the online blood-brain barrier prediction (BBB) server [17] (http://www.cbligand.org/BBB/). Higher values are predicted to pass the blood brain barrier. Size of circles = mean of -log10(e-value) for SEA-predicted targets. Larger circles indicate stronger confidence of predicted targets. Color of circles = Mean of max T values. Warmer colors (red) indicate better molecule-molecule matching, a second measure of prediction confidence. Y axis reference line (0.0815) and X axis reference line (17) are the average value for the world’s most widely prescribed psychiatric medications (aripiprazole and quetiapine).[74] These predictions suggest that nuciferine and its metabolites (O-nornuciferine, lirinidine) may be responsible for the psychotropic effects reported in humans. Chemical structures of nuciferine, O-nornuciferine, and lirinidine provided.
Next we compared the polypharmacological profile of nuciferine with two atypical antipsychotic drugs (clozapine and aripiprazole) and the typical antipsychotic drug haloperidol (Fig 2).This convergence of predictions coupled with the potential utility of a compound with high polypharmacology suggested that nuciferine (Fig 1) would be the most pharmacologically interesting to investigate. Notable predictions from the SEA [13] are listed in Table 1, of which we were capable of testing 10 via the National Institute of Mental Health Psychoactive Drug Screening Program (PDSP).
Table 1. In silico and in vitro characterization of nuciferine.
| SEA Predictions (in order of confidence) | In Vitro Affinity (nM) | Functional EC50 (nM) | Function Type |
|---|---|---|---|
| Radioligand used | |||
| D1 (5.4e-32) | 752 [3H]SCH23390 | ||
| D2 (1.6e-25) | 515 [3H]N-methyl Spiperone | 65.07 | Partial agonist |
| D3 (1.3e-12) | 741 [3H]N-methyl Spiperone | ||
| D5 (5.0e-13) | (neg) [3H]SCH23390 | 2600 | Partial agonist, ~50% |
| VMAT2 (2.1e-13) | NA | ||
| SK Channel (3.8e-11) | NA | ||
| 5-HT1A (1.2e-11) | 77 [3H]WAY100635 | 3230 | Agonist |
| 5-HT5B (1.6e-7) | |||
| 5-HT7 (4.1e-4) | 49.8 [3H]LSD | 150 | Inverse agonist |
| 5-HT2A (4.1e-6) | 312 [3H]Ketanserin | 478 | Antagonist |
| Unpredicted Hits | |||
| 5-HT2B | 41 [3H]LSD | 1000 | Antagonist |
| 5-HT2C | 60.5 [3H]Mesulergine | 131 | Antagonist |
| 5-HT6 | 268 [3H]LSD | 700 | Partial agonist, 17.3% |
| D4 | 1387 [3H]N-methyl Spiperone | 2000 | agonist |
Receptor targets are listed with their respective SEA prediction value (if available), followed by their competition binding affinity value (if available), followed by their functional EC50 value (if available) and the corresponding function type.
Information about cell lines for all assays can be found at http://pdsp.med.unc.edu/pdspw/binding.php.
The PDSP in vitro affinity screening (Tables 1 and 2) revealed a total of 13 receptors with affinities less than 1 μM, and 21 receptors with affinities less than 10 μM. SEA successfully predicted 7 out of 13 G protein-coupled receptors that were determined by the PDSP to have Ki values of less than 1 μM. Functional studies (Table 1) indicate that nuciferine shows appreciable potency as a D2 partial agonist (EC50 = 64 nM), as a 5-HT7 inverse agonist (EC50 = 150 nM), and as a 5-HT2C antagonist (IC50 = 131 nM). [30] Nuciferine was a partial agonist at D2 receptors with an activity (Emax = 67% of dopamine, Fig 4A) similar to aripiprazole (Emax = 50% of dopamine).[19, 31] In line with its partial agonist activity, nuciferine inhibited dopamine-induced activation of Gi (Fig 4B) with a potency similar to clozapine (nuciferine KB = 62 nM; clozapine KB = 20 nM) as determined via Schild regression analysis. [32] Also similar to clozapine, nuciferine exhibits 5-HT2A antagonist activity (IC50 = 478 nM) and 5-HT7 inverse agonist activity (IC50 = 150 nM), [33] and may effectively antagonize 5-HT6 receptors as a low efficacy partial agonist (EC50 = 700 nM, 17.3% maximal effect). [2, 34] Finally, nuciferine exhibited micromolar potency as a 5-HT2B antagonist (IC50 = 1 μM), a D4 agonist (EC50 = 2 μM), a D5 partial agonist (EC50 = 2.6 μM, 50% maximal response), and a 5-HT1A agonist (EC50 = 3.2 μM).
Table 2. In vitro affinity findings lacking functional measurements.
| PDSP Hits Without Functional Assay | In vitro Affinity (nM) Radioligand used |
|---|---|
| 5-HT1D | 518 [3H]5-CT |
| α1A | 1386 [3H]Prazosin |
| α1B | 1995.3 [3H]Prazosin |
| α1D | 818 [3H]Prazosin |
| α2A | 1153.5 [3H]Rauwolscine |
| α2B | 686.8 [3H]Rauwolscine |
| α2C | 692.8 [3H]Rauwolscine |
| β1-AR | 7149 [3H]CGP12177 |
| β3-AR | 1103 [3H]CGP12177 |
| DOR | 10000 [3H]DADLE |
| H2 | 1662 [3H]Cimetidine |
| MOR | 9549.00 [3H]DAMGO |
| V1A | 10000 [3H]Vasopressin |
| 5-HT5A | 1113 [3H]LSD |
Receptor targets not predicted by SEA but identified by in vitro competition binding assays for which functional assays are not available. The following receptors assayed in the PDSP screen had no observed binding of nuciferine. Receptors are listed with their associated radioligand in (). Sets of receptors using the same radioligand are indicated in []: [5-HT1B, 5-HT1E]([3H]5-CT), 5-HT3 ([3H]GR65630), β2-AR ([3H]CGP12177), BZP Rat Brain Site ([3H]Flunitrazepam), D5([3H]SCH23390), DAT([3H]WIN35428), GabaA ([3H]Muscimol), H1 ([3H]Pyrilamine), H3 ([3H]α-methylhistamine), H4 ([3H]Histamine), KOR ([3H]U69593), NMDA PCP site ([3H]MK801), [M1, M2, M3, M4, M5]([3H]QNB or [3H]NMS), NET ([3H]Nisoxetine), Oxytocin([3H]Oxytocin), SERT ([3H]Citalopram), Sigma 1 ([3H]Pentazocine), Sigma 2 ([3H]DTG), [V1B, V2]([3H]Vasopressin).
Fig 4. Nuciferine functional activity at the dopamine D2 receptor as measured via D2-mediated Gi signaling in HEKT cells.
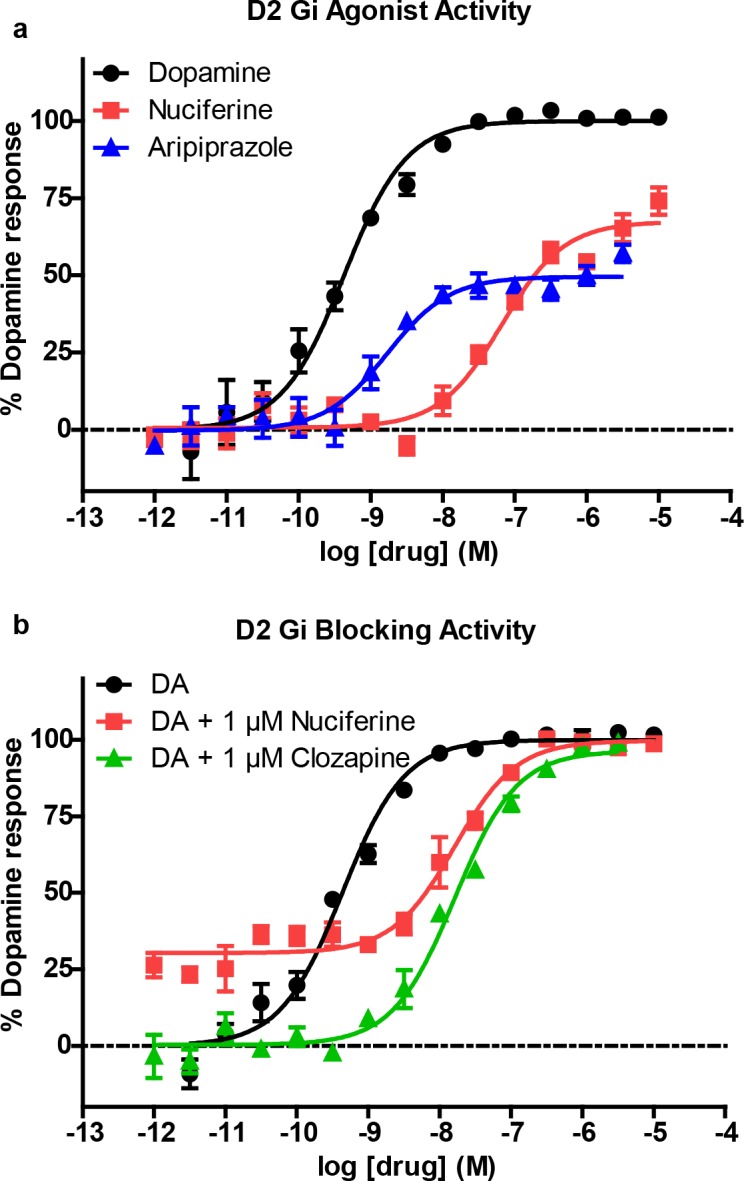
(a) Concentration-response curve of nuciferine (red) compared to dopamine (black) and aripiprazole (blue). (b) Concentration-response curve of dopamine (DA) in the presence of 1 μM nuciferine (red) or clozapine (green) showing rightward-shift of DA indicative of competitive antagonist activity. The data represent concentration-response curves of normalized data with respect to dopamine performed in triplicate (mean +/- s.e.m.; n = 3).
3.2 Assessment of aggregator properties
The predominantly low-potency antagonist activity of nuciferine suggested that nuciferine could be functioning as a colloidal aggregator, a well-known mechanism of promiscuous activity in early discovery, such as high-throughput screening. [35, 36] Recent studies have shown this mechanism can affect membrane-bound receptors such as GPCRs [37] in cell-based screening. At concentrations relevant to this study (< = 10 μM), dynamic light scattering determined that nuciferine did not scatter light above background in aqueous buffer, suggesting colloidal aggregates of nuciferine are not formed at this concentration. Consistent with this, nuciferine did not inhibit AmpC β-lactamase at these concentrations; AmpC is a counter-screening enzyme widely used to test for colloidal aggregation [35, 36, 38] (data not shown). The lack of particle formation by light scattering and the lack of inhibition of the orthogonal counter-screening enzyme supports the idea that nuciferine is not acting promiscuously via colloidal aggregation in the assays described here.
3.3 Dopamine transport by DAT and VMAT2
SEA predicted that nuciferine may interact with the vesicular monoamine transporter-2 (VMAT2). To determine if nuciferine affects uptake at VMAT2, we conducted dopamine uptake experiments in vesicles isolated from HEK cells overexpressing VMAT2 and found no effect of nuciferine on DA uptake (Fig 5A). We also tested whether nuciferine affected uptake in whole-cell uptake studies in HEK cells expressing DAT and VMAT2 or DAT alone as a control, as described elsewhere. [39] In whole cells expressing DAT and VMAT2, the total capacity of the cell to take up and store dopamine is measured. This has a DAT-mediated and a VMAT2-mediated component, as previously demonstrated.[39] Comparison of the effects of compounds in these two cell types, as well as in isolated vesicles, aids in separating out these two components of uptake in whole cells. Nuciferine pre-treatment had no effect in HEK-DAT/VMAT2 cells (Fig 5B). Counter to predictions, in the HEK-DAT control cells, nuciferine pre-treatment increased uptake in HEK-DAT cells by 60% over the vehicle control (Fig 5C; EC50 = 1.8 nM).
Fig 5. DAT modulation by nuciferine.

(a) Concentration-response curves of nuciferine on vesicular uptake in isolated vesicles, (b) concentration-response curves of nuciferine on vesicular uptake in HEK cells transfected with DAT and VMAT2, (c) concentration response curves of nuciferine on vesicular uptake in HEK cells transfected with DAT. Data are presented as a percentage of the response to vehicle control. The data represent concentration-response curves of normalized data with respect to vehicle performed in triplicate (mean +/- s.e.m.; n = 3).
3.4 DOI-induced head-twitch response (HTR)
Mice pretreated with nuciferine (1.0, 3.0 and 10.0 mg/kg, i.p. or 3 mg/kg, s.c.;15 min prior to DOI) showed a dose-dependent inhibition of the head-twitch response produced by 1.0 mg/kg DOI during the course of testing (Fig 6A). Bonferroni corrected pair-wise comparisons indicated that regardless of route of injection, nuciferine attenuated DOI-induced head-twitches at 10, 15 and 20 min (ps<0.05). When the different doses of nuciferine were examined, a RMANOVA revealed a significant effect of time [F[3,36] = 232.725, p<0.0001], and a significant time by treatment interaction [F[9,36] = 16.170, p<0.0001]. Bonferroni comparison results indicated that 3 and 10 mg/kg nuciferine decreased head-twitches at 5, 10, 15 and 20 min (ps<0.05) relative to DOI-treated mice. Differences in responses to the 3 and 10 mg/kg doses were not statistically significant (Fig 6A). By comparison, 1 mg/kg nuciferine exerted no statistical effect on the DOI induced head-twitches.